
 杏耀资讯
杏耀资讯  杏耀注册
杏耀注册  杏耀登录
杏耀登录  杏耀平台
杏耀平台  杏耀代理
杏耀代理  杏耀APP
杏耀APP  杏耀加盟
杏耀加盟  杏耀招商
杏耀招商  杏耀开户
杏耀开户  网站首页
网站首页如何理解化学势的物理意义?
自学统计物理和固体物理的时候总是会碰到化学势,虽然其涉及的基本数学推导过程都已经熟悉,但仍然对它的物理意义困惑,它常见的表达式是吉布斯自由能对成分的偏微分,又可以由巨正则分布公式得到它是引入一种成分后系统总能的变化,但却总不能像常见的能量,温度之类物理量一样将其深入理解,也仅仅只能将它当做类能量物理量将其处理,不知道有没有更直观深刻的理解方法? @傅渥成@何史提@Zeldovich Yakov@Tianqi Chen
从定义上看,“化学势”和粒子数有关,具体来说,是粒子数目的变化导致的Gibbs自由能的变化。但这还是不能更直观的理解它到底是什么。好,在具体的例子中,特别是涉及到粒子数目变化的例子中理解“化学势”,会很直观。
最简单的例子是:单一物质处于气-液两相共存态(vapor-liquid coexistence)
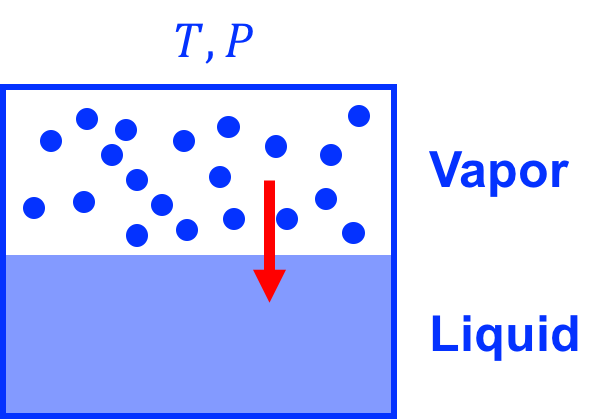
在这种共存态中,个粒子处于气态,
个粒子处于液态,并且满足
。各自的Gibbs自由能表达式为
和
。假设某一时刻有
个粒子从气态进入了液态,并且
,
。这必然会导致气-液共存系统的Gibbs自由能发生变化:
利用Taylor展开:
(I)对于稳定的气-液共存态,系统Gibbs自由能对于的扰动应当变化为0,因此:
(II)如果 ,那么
,也就是说分子从气态进入液态是允许的。
(III)如果 ,那么
,也就是说分子从气态进入液态是不被允许的。
从上面的讨论就可以看出来:
气-液平衡的条件是气态的和液态的
相同;
对于未平衡的体系,分子是从具有较高的相流向
较低的相。
这个,就是化学势。
其实,系统中存在“化学势”的差异,就是一种驱动分子在不同相之间转移的动力,最终使系统中各处的“化学势”处于相同,即系统达到稳定。
溶液中的溶质存在浓度差异时,粒子会从浓度高的区域流向浓度低的区域。对于理想溶液,组分的化学势确实正比于浓度的对数;对于简单的理想气体模型,化学势正比于密度的对数。这些都表明,“化学势”的差异,驱动了分子在系统内不同相之间的“流动”。
考虑到,大概可以把化学势理解为在维持总能量
和能级
不变的情况下,衡量体系粒子数
和混乱程度
之间关系的物理量?
在不变的情况下,
不可能和
没有关系,那么就把它们之间的依赖关系用
来描述?
---------------------------------------------------------
突然想到了温度的定义。那么就试着从热力学的角度给一个理解吧。
这里化学势的定义和温度的定义也是很类似的(温度是什么实际上并不是很显然的问题)。热力学第零定律告诉我们有一种叫做热平衡的状态,达到热平衡的两个物体之间不交换热量,这样的话我们就可以给每个物体贴上一个叫做温度的标签(规定温标)满足热量只从这个标度高的物体向这个标度低的物体转移,而在两个标度相同的物体之间不发生转移。
同样,对于化学势,我们考虑两个可以相互交换粒子的系统。实验上发现,两个系统在某种情况下相互交换粒子的宏观表现为0,达到动态平衡,我们就可以说这两个系统必定有某个量相同,并把这个强度量称为化学势。
推而广之,对于任何一个广延量X,和两个可以“交换”X的系统,都会在某种情况下达到动态平衡,也就对应着两个系统此时拥有的一个相同强度量Y。在X是热量的情形下,Y是温度,X是体积的情形下Y是压强,X是粒子数是Y就是所谓的化学势了。
上面的定性分析实际上并没有给出化学势的具体公式。实际上如果只考虑单一广延变量的情形,作为衡量强度的标度并不是唯一的。比如给定了一个温标T,那么任何关于T的单调递增函数f(T)都可以作为温标(化学势同理)。但我们总是希望找到一些好用的衡量强度的标度,一个比较有效的办法是将同能量联系起来。于是就有了
。剩下的就是算算算了。
两个子系统间存在能量交换,那么可以定义一个量温度,温度相等时两边平衡。
如果两个子系统之间除了能量交换还能进行体积交换(比如两部分气体中间连一个活塞),那么就算温度相同也不一定平衡。我们可以再定义一个量叫压强,压强相同时两边就平衡了。
如果两个子系统还可以交换粒子数,那么相应的可以再定义一个东西。这东西就叫化学势。
化学势本身可以理解为一种广义力,对于一个电荷体系,假定整个体系的电势能为: ,
表示位置,则该体系受到的静电作用力为:
(
表示该体系的电势能V对位置x求梯度),
倾向于做功使体系改变其位置,以降低体系的电势能,体系平衡的条件为:
。
同样的,对于一个共有a种组分的化学体系,不同物质组分的物质的量为 ,体系的自由能为
,则可定义驱动化学反应的广义力为:
(
表示该体系的自由能G对该化学体系的物质组分求梯度),广义力
倾向于做正功,改变体系的物质组分,以降低体系的自由能,使体系的能量贬值,保证体系的化学反应服从热力学第二定律。很显然的,平衡的条件为:
,即体系的总化学势=0,系统广义上“受力平衡”。
在第一部分,为了对化学势进行解释,引入了自由能的概念。于是,很自然地会产生这样一个问题:自由能是什么?为什么要引入这样一个概念?这一部分便着重回答这个问题。
对于化学反应体系本身,整个化学反应从开始到结束,体系总的熵变为:
,
其中: 为可逆性熵流,表示体系在化学反应中因与外界交换物质与能量而流入体系中的熵;
为不可逆性熵产,表示因化学反应本身的不可逆性而“凭空”产生的熵。
热力学第二定律要求: 必须
,即任何化学反应都是热力学不可逆过程,必须有新的熵“凭空”产生。
于是,对于等温等压条件下的封闭容器中(若是其他条件则需要另外讨论)的化学反应,根据能量守恒定律:
有 ,其微元关系为:
(注意:此处
表示微元,
表示全微分,两者含义不同。因为
不是状态量,严格来讲不存在全微分。以下,凡是不存在全微分的量均用
表示,存在全微分的量则用
表示)
即:体系吸取的热量=系统的焓变。焓就可代表系统的能量。
对于等温等压条件下的封闭容器,唯一与外界环境发生关联的是体系吸取的热量 ,所以体系的可逆性熵流
必然蕴含于 吸收的热量
之内。关于这两者,存在如下关系:
,
这一表达式的含义是:体系在从状态 变化到状态
的过程中,只要吸收外界热量就会使得外界的熵流入体系内。
将体系焓变 的表达式带入
的表达式中有:
;其微元关系为:
移项得: ;
很明显地, 这一项具有能量量纲。它与体系不可逆性熵产
相互关联。前面提到:
表示因化学反应本身的不可逆性而“凭空”产生的熵,是热力学第二定律的体现。而热力学第二定律造成的结果就是能量贬值:原本“自由的”容易利用的能量,转变为了“封闭的”难以利用的能量,降低了体系对外做功的能力。
因此,在准静态过程的假设下,可以定义新的状态函数——自由能: ,
表示体系中未贬值的能量;
因此: 表示:体系因为发生了热力学不可逆过程(化学反应)而被热力学第二定律强行贬值的能量,化学反应的后果就是使得体系的可用能量(自由能)减少,体系做功的能力降低。
有了以上的分析,让我们再次回到化学势的概念上来,第一部分提到:化学势是一种广义力 。则
做的功可以表示为:
,即:广义力
做功会使得体系的自由能降低,使得体系的能量发生贬值,“自由”可用的能量减少,体系“凭空”产生了新的熵。这是热力学第二定律在化学反应中的体现。即:化学反应是广义力
做正功的结果,也总是向着广义力
正功的方向进行。当广义力
时,反应达到平衡态,广义力不能再继续做功了,体系对外做功的能力为0。
另外,需要注意的是,以上的推导均是在“等温等压条件下的封闭容器中的化学反应”这一前提下进行的,若化学反应发生于其他条件之下,则 、
、
、
等物理量的表达式将发生变化。相应的广义力
也需要重新定义。
在第二部分, 被直接 称为自由能,而非吉布斯函数或者吉布斯自由能。这是因为:自由能与吉布斯函数并非始终等同。为了描述清楚两者的关系,重新约定:
为体系的自由能;
体系的吉布斯函数;
在热力学中:
体系的焓定义为: ;
体系的吉布斯函数定义为: ;
对 与
进行全微分有:
在既不等温,也不等压的封闭容器中的化学反应,依旧有能量守恒关系:
(若容器为开放容器,则
。此时还需要考虑与外界的物质交换所造成的熵流。熵流本身可正可负,为正则表示环境中的熵流入体系内部,为负表示体系中的熵流出到环境中);
于是: ,将之带入吉布斯函数的全微分式中,有:
第二部分提到: ,最终可以得到,对于既不等温,也不等压的封闭容器中的化学反应,有:
因此在既不等温,也不等压的条件下,封闭容器中的化学反应体系的 ,而是相差了
这两项,吉布斯函数中广义力
所做的功被体系温度和压强的变化所“遮盖”。此时,
不再是热力学第二定律的体现,也就不能用于化学反应方向的判定。
只有在等温等压封闭容器的条件下( 且
),才有
。
在这个部分,依旧约定:
为体系的自由能;
体系的吉布斯函数;
对于一个实际的化学反应: ,在发生化学反应时,在
时间内,A、B、C、D四个组分物质的量的变化,可以写成一个向量:
为了讨论方便,引入化学计量数向量: ,约定反应物的化学计量数为负,生成物的化学计量数为正。并引入反应进度
的概念,定义:
于是:
;
第二、三部分提到,热力学第二定律始终要求: ,因此自然有
,于是就得到了热力学第二定律在化学反应中的描述:
。
于是在计入化学计量数约束的条件下:
表示化学反应自发正向进行;
则表示化学反应达到了平衡态(不同于无约束条件下的
的条件);
表示化学反应正向进行时会违反热力学第二定律,于是化学反应只能自发反向进行。
同样的,在等温等压的封闭容器中,有 。
化学势在热力学里非常容易理解,就是Gibbs自由能对原子数(视为实数)的偏导数。根据推导,还可以推出其另外三个等价定义。关于化学势的应用,朗道给了许多有意思的例子。
简单稀溶液理论统计物理中要注意,用巨正则系综是无法推导出来化学势的,化学势在巨正则系综中是个自变量,可以随意取值。只有在正则系综中可以得到。但是对于粒子数可变的系统,从正则系综出发,又难以推导相关配分函数。只好用巨正则系综推导配分函数、再用拉普拉斯逆变换到正则系综、再计算化学势的曲折办法。Am. J. Phys.有一篇B. McCoy写的文章,详细介绍了这套计算,并用Mathematica算出了费米子、玻色子理想气体的化学势随温度变化。
理想气体化学势随温度的变化做分子动力学、萌卡、相场计算的话,化学势也很容易理解,可以用模拟计算得到的数据获得化学势信息。phase field计算专家陈龙庆老师有一篇文章,从热力学角度讲解化学势,简单直观。
https://www.cambridge.org/core/journals/mrs-bulletin/article/chemical-potential-and-gibbs-free-energy/2BACF973D95A697C947E741630951F46#但到了“量子多体理论”中,它的出现就非常神秘了。我旁听同事讲的QMB课程,似乎是由Lagrange乘子法引入了一个参数,这个参数就叫做化学势。在BEC相关的计算中,就这么跳出来了。更加“物理”的讲解我还没见过。也是我QMB、半导体物理相关文献读的少,无法多谈。希望各位专家能深入谈一下。 @浅斟低唱 @Narayan @kBlnW

最新更新
猜你喜欢
关注我们


